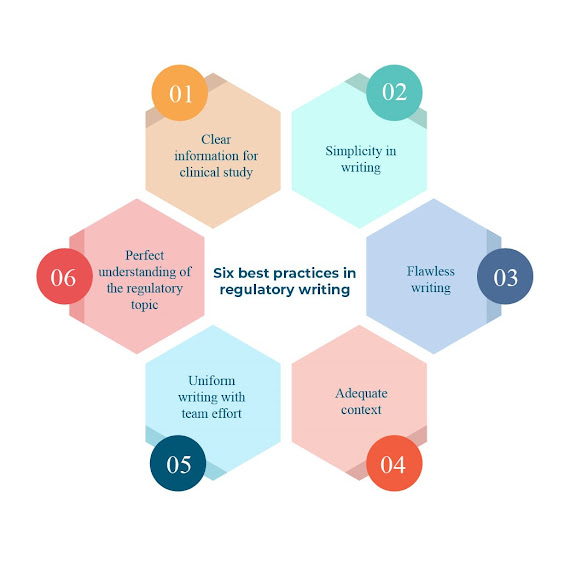
REGULATORY WRITING – VITAL FOR CLINICAL STUDY

Regulatory writing, a branch of medical writing, is essential for the conduction of clinical trials approved by the higher regulatory authorities. Regulatory medical writing encompasses various clinical documents produced during the various phases of treatment starting from description and documentation of the clinical trials to preparation of the regulatory suggestion documents. Examples of clinical documents applied in regulatory writing are the Common Technical Documents (CTD), Clinical Study Reports (CSRs), Clinical Study Protocols (CSPs), Patient Narratives and Safety Reports. The important national regulatory authorities are: · Central Drugs Standard Control organization (CDSCO) (India) · Food and Drug Administration (FDA) (United States) · Health Canada (Canada) · European Medicines agency (EMA) (European Union) · Ministry of Health, Labour and Welfare Japan (Japan) The efficacy and the safety of the drugs for overall cl